Research
Our objective is to improve the drug discovery and safety pipeline, our understanding of sequence-structure-function relationships, often from an evolutionary perspective, and broadly contribute open data and methods to further our collective understanding of living systems. Along the way, we work to improve access to knowledge through open scholarly communication.
What is an ‘urfold’?
We develop new deep learning algorithms such as Autoencoders to learn local substructures of geometry and biophysical properties to help understand this concept of ‘Urfold.’
| We suspect that there is a level of granularity of protein structure intermediate between the classical levels of “architecture” and “topology,” as reflected in such phenomena as extensive three‐dimensional structural similarity above the level of (super)folds. We examine this notion of architectural identity despite topological variability, starting with a concept that we call the “Urfold.” We believe that this model could offer a new conceptual approach for protein structural analysis and classification: indeed, the Urfold concept may help reconcile various phenomena that have been frequently recognized or debated for years, such as the precise meaning of “significant” structural overlap and the degree of continuity of fold space. More broadly, the role of structural similarity in sequence↔structure↔function evolution has been studied via many models over the years; by addressing a conceptual gap that we believe exists between the architecture and topology levels of structural classification schemes, the Urfold eventually may help synthesize these models into a generalized, consistent framework. | 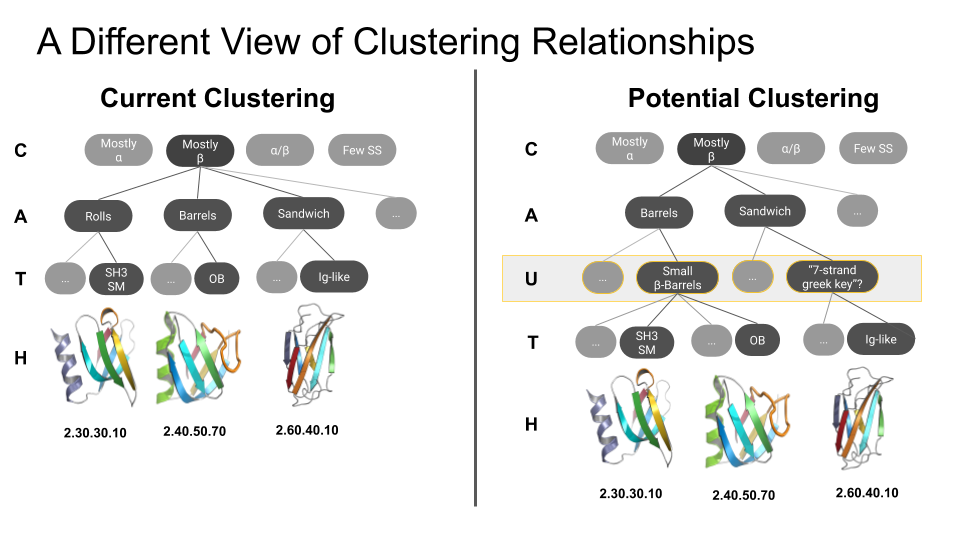 |
More Info | DeepUrfold: Stochoastic Block Model Community Analysis
Protein Interaction Prediction
We write deep learning models to better predict protein•protein interactions (PPIs). Protein•protein interactions (PPI) mediate biological functions and are crucial to understanding cellular pathways. Despite the molecular crowding in the cell, it is remarkable that two proteins, A and B, find each other to form either transient or permanent interactions (two distinct interaction types). Current methods for predicting interaction partners and residues involved in binding sites are of only limited efficacy because they (i) are trained on small datasets, (ii) ignore binding affinities (interaction types), and (iii) ignore domain-level and superfamily information. Also, predictions generally remain unverified. We hypothesize the including binding affinities will be important because the features from transient or permanent will be sufficiently different; if models do not specify interaction type, they may learn generalized binding site features of all, not specific to either type, missing many potential candidates. We seek to devise a new PPI benchmark that is at the domain/superfamily level, incorporates known binding affinities, and contains representative features for all atoms. In summary, we will develop a methodological pipeline for validating inferred interactions in order to create a larger dataset, organized by binding affinity classes and protein superfamilies. We will also use unsupervised deep learning methods to perform feature selection and standardization. This will serve as a useful community resource for deep learning of protein interactions.
Computational Systems Pharmacology
With the accomplishment of the human genome project and the progress of the genome-wide association study(GWAS), more human diseases’ targets, signaling pathways, and pathological mechanisms are being elucidated. These advances propel more new drugs to be quickly designed and applied to meet clinical requirements. Computational system pharmacology (CSP) has been recognized as an effective way for drug design and discovery by utilizing omics data. We will use the CSP strategy to (1) develop novel methods and (2) apply in-house/sophisticated technologies to facilitate the launch of more new drugs. (1) We will develop new drug screening skills, drug target prediction method, drug polypharmacology techniques by combining traditional docking concepts, umbrella sampling, machine/deep learning models et al. (2) We apply the in-house/sophisticated approaches such as docking, high-throughput/content screening, and binding free energy calculation for specific drug design and discovery, drug repurposing, and drug mechanisms of action to treat different intractable diseases. Currently, our projects and research involve different diseases including Alzheimer’s disease, Malaria, non-small cell lung cancer, melanoma, and leukemia.